

In a study published this month in the Proceedings of the National Academy of Sciences, Dr. Amantha Thathiah, assistant professor of neurobiology at Pitt, applied her background in cancer research to developing a cure for Alzheimer’s disease.

Amantha Thathiah
Thathiah and her group focused on a family of proteins called G protein-coupled receptors, or GPCRs, that are widely present throughout the human body and play important roles in physiological and pathological conditions, from learning and memory formation to cancer progression. The scientists reasoned that these proteins’ unique ability to preferentially modify, or “bias,” their activity based on signals they receive from their environment could be harnessed to develop new drugs for Alzheimer’s disease.
Given the involvement of GPCRs in numerous biological processes, approximately 40% of all medications on the market today target GPCRs. Medications that either activate or block various classes of GPCRs are used to control blood pressure and treat heart disease, alleviate pain and manage neuropsychiatric disorders, among other conditions. However, despite their presence in multiple cell types comprising the brain tissue – from neurons to resident immune cells – the role of brain GPCRs in Alzheimer’s disease has not been widely studied.
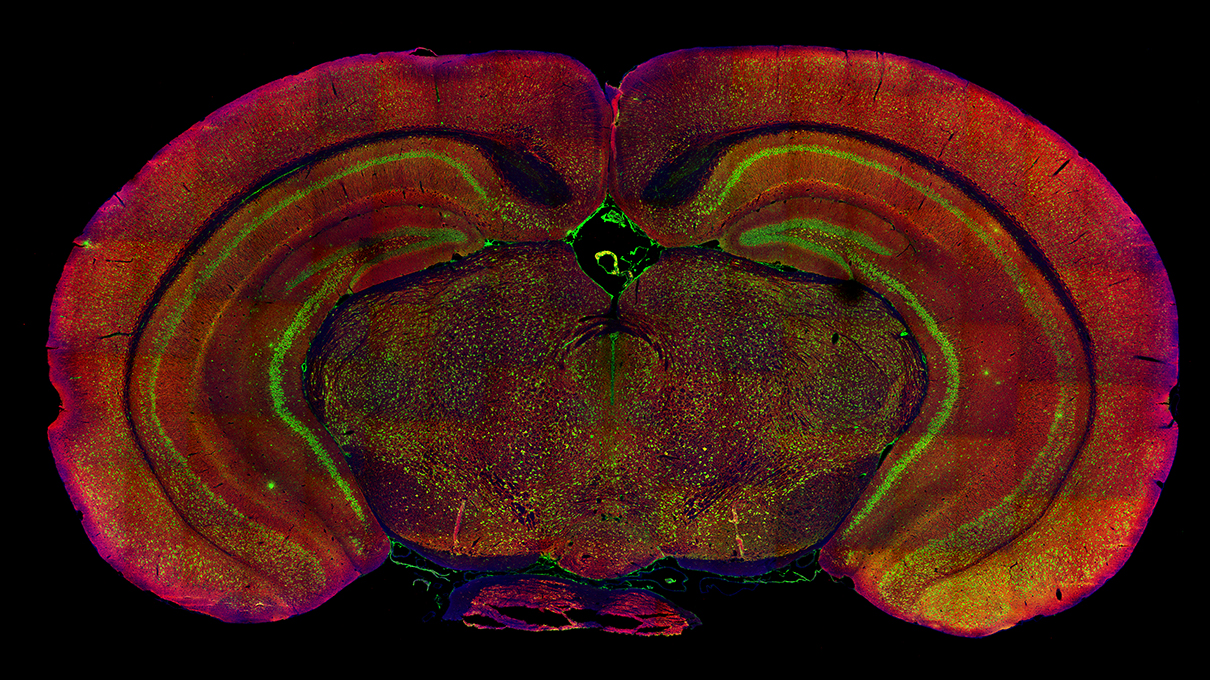
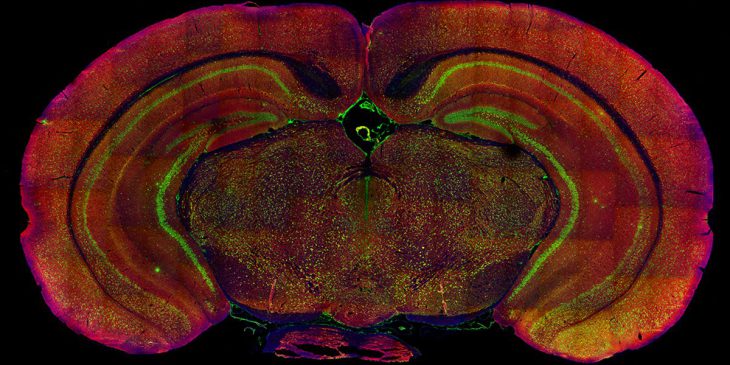
Immunofluorescent staining of the mouse brain
Part of the reason is the complexity of GPCR activation. When GPCRs are activated, they can recruit small molecule messengers called G proteins or attract alternative partners called beta-arrestins, which initiate different cascades of events that can be either beneficial or harmful to the overall cell health. For example, the beneficial effects of many GPCR-targeted medications are often also associated with a list of potential side effects. Accordingly, various research groups are exploring opportunities to develop “biased” GPCR drugs that preserve the beneficial effects of GPCR activation without the unwanted side effects. However, such GPCRs have not yet been explored in Alzheimer’s disease.
Previous studies by Thathiah and her group discovered that a subset of Alzheimer’s disease patients has elevated levels of a GPCR called GPR3. They also found that altering a mouse’s genetic code and deleting the Gpr3 gene – which directs the production of GPR3 – reduces the amyloid plaque burden in the brain but elevates anxiety levels associated with Alzheimer’s disease.
“Simply put: Targeting this protein in Alzheimer’s disease holds promise, but merely removing it isn’t enough,” said Thathiah.
So, the researchers turned to the idea of exploiting the ability to bias GPR3 activation to preserve the beneficial effects while avoiding the detrimental effects associated with the complete elimination of GPR3 signaling.
Mice with an inactivating mutation in Gpr3 that prevented beta-arrestin binding did not display the signs of elevated anxiety or memory impairment associated with a complete deletion of Gpr3. Excitingly, these animals also had lower amyloid plaque burden, which was accompanied by the activation of the brain’s protective immune response.
As a whole, the study suggests that developing drugs that exploit biased GPCR signaling has the potential to become a safer therapy for Alzheimer’s disease and numerous other conditions. While the scientists still have a considerable way to go to turn their discovery into a product on pharmacy shelves, they are optimistic.
“Research studies, guided by funding agencies, tend to focus on avenues of research that are primarily ‘under the streetlamp.’ Perhaps the most transformative discoveries will require us to start looking in the shadows at less described avenues for targeting the disease,” said Thathiah. “Our data provide proof-of-concept that biased GPCR signaling is a potentially viable avenue for therapeutic intervention in Alzheimer’s disease.”
Related Posts:






![]()
![]()
![]()
![]()
![]()
![]()
![]()